서 론
의약품 사용에 따라 나타나는 이상사례(adverse event, AE)는 예측과 적절한 정보제공으로 일정부분 예방이 가능하므로, 약물 이상반응 발현 빈도를 감소시키고, 환자 치료의 질을 향상시키며, 의료비를 절감시킬 수 있도록 약물 이상반응 관리의 중요성을 인식하고, 이를 위한 노력이 필요하다[1]. 의약품 허가 당시에는 유익성(benefit)이 위해성(risk)보다 크다는 것을 입증하여 허가를 받게 된다. 새로 개발된 약물이 시판 허가를 받으면 많은 수의 환자들이 장기간에 걸쳐 복용하게 되므로 시판 전 임상시험에서 발견되지 않았던 드물지만 중대한 유해반응들이 나타날 수 있다. 시판허가를 받기 위한 임상시험은 가능한 빨리 허가를 받기 위해 관찰기간이 제한되고, 연구 대상자의 수가 한정되어 드물게 발생하는 유해반응을 관찰하기 어렵다. 따라서 시판 후에도 대규모 인구집단을 대상으로 장기간에 걸친 관찰을 통하여 그 약물의 안전성을 충분히 확인하는 것이 필수적이다. 허가 당시 얻을 수 있는 유익성과 위해성 정보의 양과 질에는 한계가 있고, 시판 후 조사 및 보고를 통한 보다 실질적이고 새로운 정보가 지속적으로 수집되면 추가되는 정보로 인해 유익성과 위해성의 균형이 변화하게 되므로 의약품의 전주기를 통해 일정 시점에서 유익성과 위해성을 지속적으로 비교 평가할 필요가 있다[2].
미국, 일본, 유럽이 주축을 이루는 국제의약품규제조화위원회(International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use, ICH) [3]에서는 의약품을 보다 안전하게 사용하고, 평가하기 위한 과학적인 노력을 계속해 오고 있다. 의약품의 시판 전 임상시험, 안전성 및 유효성과 품질관리를 위한 가이드라인을 지속적으로 발간하고 시판 후 수집되는 안전성 정보수집·분석 및 관리뿐만 아니라 시판 전후를 통합한 전주기 유효성·안전성 정보통합분석을 위한 방법론 및 가이드라인을 여러 규제기관, 국제의학단체협의회(Council for International Organizations of Medical Science, CIOMS) 등 전문가들과의 공동연구 및 합의를 통하여 발간해 왔다. E2C (R1) (임상적 안전성 정보 관리: 시판의약품의 정기적 안정성 정보 보고)의 후속으로 2012년 E2C (R2) (임상적 안정성 정보 관리: 유익성-위해성 평가 정기 보고)가이드라인이 도입, 시행되면서 허가된 의약품의 유익성에 비추어 새로운 안전성 정보를 평가하도록 하였다. 또 E2E (의약품 감시계획)의 도입 후 규제당국과 기업은 의약품의 개발·신청 및 허가 심사단계에서부터 안전성에 대한 개입을 검토하도록 하여 의약품의 유익성에 관하여 중요한 새로운 위해성 정보에 대한 의미 있는 평가가 필요하다는 인식이 점점 커지고 있다. 이러한 의약품 부작용 예방 및 저감화 등 안전관리체계 운영에 있어 국내외 개별 이상사례 보고·수집의 전자화 및 표준화는 필수적인 요소로 인식되고 있다.
기술적인 면에서 의약품 부작용은 ICH 지역에서 E2B (임상적 안전성 정보 관리: 개별 사례 안전성 보고의 데이터 요소) 가이드라인[4] 도입을 통한 개별 이상사례 보고(Individual Case Safety Report, ICSR)의 전자수집과 자동 데이터마이닝(data mining) 기술이 발달하게 되면서 빠르고 정확한 실제 의료현장(real world)에서의 자료수집과 모니터링이 가능하게 되었다. 또한 빅데이터(big data) 처리 및 분석기술 등 정보통신 기술의 발달로 전자의무기록(electronic medical records, EMR) 및 보험청구 자료 등 다양한 출처의 의료정보를 활용하여 근거에 기반한 의사결정에 도움을 주고 있다. 표준을 적용한 데이터의 정규화는 빅데이터 기술을 적용한 자료의 분석과 주기적 인상시 모니터링 시스템 구축을 위한 필수요건이라 할 수 있으며, 국외에서는 이미 이전부터 이러한 정보통신 기술을 활용하여 의약품 부작용 보고서식 전자표준으로서 ISO/HL7 27953 표준을 개발하였다. 또 ICH는 표준개발기구(Standards Development Organization, SDO), CIOMS 등 다른 표준과의 협력작업을 통하여 의약품 안전성 정보분석을 위하여 표준용어집인 의약품 안전관리 수행을 위한 국제의약용어집(Medical Dictionary for Regulatory Activities (MedDRA), ICH M1)과 규제정보의 전송을 위한 전자표준(Electronic Standards for the Transmission of Regulatory Information, ESTRI: M2, E2B 및 M8)을 개발해 왔다[5]. 이 국제의약용어와 전자표준은 국적·지역에 국한되지 않고 전세계적으로 퍼져 있는 여러 이해관계자 간의 국제적인 자료 교환과 통합분석을 그 일차적인 목적이라 할 수 있으며, 나아가서는 의약품 전주기에서 수집된 다양한 정보를 정규화(coding)하여 여러 데이터베이스(Database, DB) 각각의 자료는 물론 이들로부터 추출·변환·통합된 새로운 DB자료의 통계적 분석을 기술적으로 가능하게 한다. 이를 통하여 지속적이고 주기적으로 의약품의 유익성-위해성 균형평가(benefit-risk balance assessment, BRBA)를 수행하고, 이를 위해성 관리계획(risk management plan, RMP)에 반영할 수 있다.
우리나라는 2016년 11월 ICH에 가입승인되면서 E2B와 M1 (Med-DRA)을 5년 이내에 필수 이행해야 하는 상황이며, 하루 빨리 국내 부작용보고시스템에 E2B 가이드라인을 적용할 필요가 있다. 우리나라는 비교적 이른 시기에 전자정보통신기술의 발전을 이루어 앞서 언급한 바와 같이 의약품부작용보고에 있어서도 IT 기술을 적용하여 보고시스템을 개발하였다. 그러나 국내 시스템개발 당시에는 CIOMS Form I에 맞추어 전자보고를 위한 시스템이 개발되었다. 이 시스템에는 WHO-ART, KCD-6 등 제한된 어휘(용어)가 일부 적용되었으나, 보고양식에 대한 국제적인 전자표준은 적용되지 않았고, 사용자의 필요성에 따라 유지·관리되어 왔다. 국내에서는 지금까지 자발적 부작용보고자료를 이용하여 실마리 정보검색과 국외 안전성정보모니터링 등 모니터링 수준에서의 약품안전관리를 수행해 왔으나, 최근에는 위해성 관리계획(RMP) 제도(2015) 시행과 품목허가갱신 제도(2016)를 도입하여 국내에서 발생할 수 있는 한국인 특이적 부작용 탐색과 이 정보를 활용한 선제적 부작용 안전관리를 위한 제도적 기반을 마련하고, 시판 안전관리를 강화하고 있다. 이는 허가 당시 얻을 수 있는 정보의 질과 양에 대한 한계를 극복하고, 의약품 부작용에 대해 사전예측을 통한 예방 및 관리체계로 전환하는 계기가 될 수 있다. 기술적으로는 빠르게 누적되는 부작용 데이터를 효율적으로 수집하고, 자동화되고 검증된 분석절차를 마련하여 자료를 분석할 수 있으며, 이 자료로부터 도출된 안전성 정보의 신뢰성을 높일 수 있게 되어 국내 부작용 분석의 과학적 수준을 향상시킬 것이다. 또 한국제표준의 도입은 국제보건기구협력센터인 WHO-UMC 및 다른 규제기관과 관련 업계 등 이해관계자 간의 정보 전자전송을 효율적으로 수행하는 데 도움이 될 것이다. 따라서 매우 빠른 속도로 증가하는 부작용보고자료의 효율적인 수집관리 및 분석을 위한 전자표준 도입이 시급하며, 안전성 정보수집을 강화할 필요가 있다.
본 론
ICH E2B (R3) 가이드라인
목적 및 배경
E2B (R3) 가이드라인 구현의 목적은 다른 ICH 가이드라인과 마찬가지로 보고의 출처 및 전송 대상에 관계없이 모든 종류의 ICSR의 전자적 전송에 대한 모든 데이터 항목의 정의를 표준화하는 것이다. 이 가이드라인은 허가 전 또는 허가 후 모든 기간에 ICSR에 관한 데이터 항목에 대해 기술하고, 이상반응 및 이상사례보고에 모두 적용된다. 이 가이드라인의 기술적인 목적은 전송가능한 ICSR 메시지를 구성하는 구현 시스템에서 보고자 및 수신자(제약업체, 규제당국 및 비영리 스폰서 포함)를 지원하기 위한 것이다. ICSR의 표현은 특정 플랫폼, 응용프로그램 및 시스템 개발업체 등에 의존하지 않고 국제표준을 준수해야 한다.
적용대상
적용범위에 있어서 이 가이드라인의 ICSR은 형식과 내용면에서 규제당국을 포함한 많은 이해관계자 간에 의학적 내용이 정확하게 보고되도록 많은 데이터 항목으로 구성되어 있다. 따라서 표준의 신뢰성과 유용성을 유지하기 위해서는 추가적인 로컬 데이터를 포함할 필요는 없으며, 가능하면 이러한 정보의 입력을 피하는 것이 좋다. ICH E2B (R3) ICSR을 위한 이 가이드라인의 적용 범위에 데이터베이스 구조의 정의, 서면보고양식의 디자인, 품질관리/품질보증의 관점 또는 기술적인 안전성 문제 등은 포함되지 않는다. 국내 및 국제협정이나 규정 및 환자의 안전보호의 관점에서 국제사회의 구성원으로서 정보원에서 규제당국 및 제약회사로의 정보교환, 규제당국 간의 정보교환, 제약업체와 규제당국 간의 정보교환, 제약업체 간의 정보교환, 임상시험 책임자에서 임상시험 의뢰자를 거쳐 윤리위원회로 정보교환, 규제당국에서 세계보건기구(World Health Organization, WHO), 국제 의약품 모니터링 협력센터(Collaborating Center for International Drug Monitoring)로 의 정보교환과 같은 안전성 정보(예: ICSR) 교환에 적용될 수 있다.
기술적내용: ICSR 세부항목설명
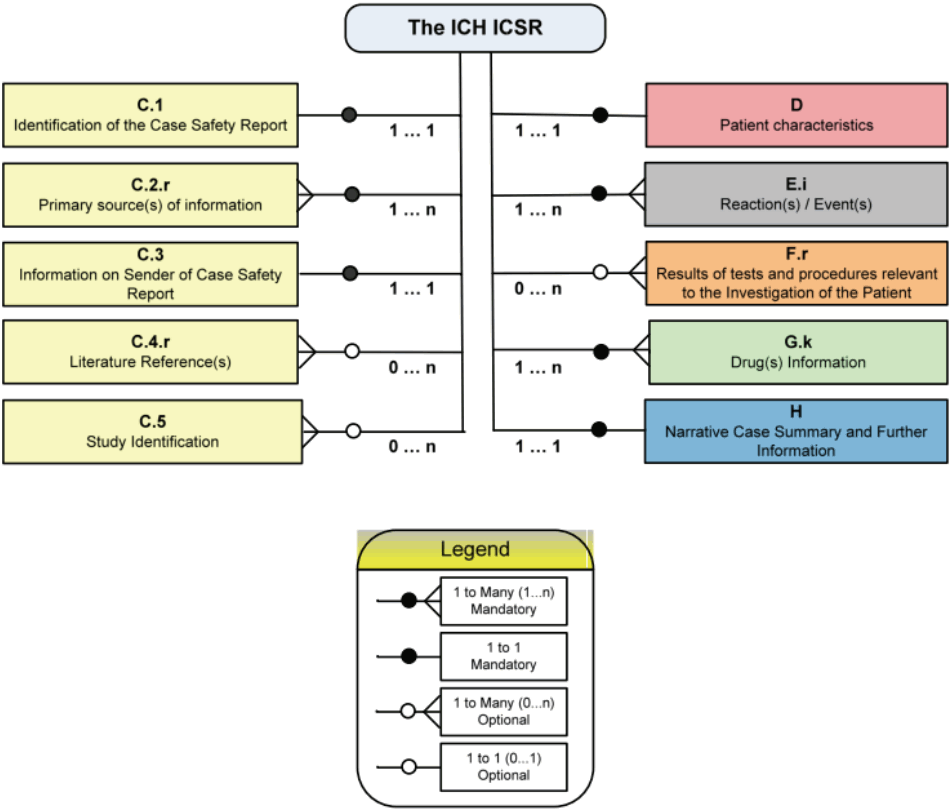
E2B (R3)에 기술되어 있는 실무 요구사항을 지원하는 소프트웨어를 개발하려면 기능 및 절차에 대한 요구사항을 충분히 이해하고 전자적 메시지에 제대로 반영되도록 할 필요가 있다. 전자적 메시지는 데이터 항목의 정확한 정의(XML 스키마)를 포함할 뿐만 아니라 효율적인 정보교환을 위해 요구되는 데이터 항목 간의 관계를 유지해야 한다. 데이터 관련도, 속성목록, 숫자코드 및 ICH ICSR 스키마 제약의 개발은 ICSR의 전자적 전송을 촉진하는 소프트웨어 사양의 개발프로세스이다. ICH ICSR 메시지에 필요한 스키마는 ICH E2B (R3) 가이드라인의 부록 (A)에 기재되어 있고, ICH ICSR 메시지에 대한 E2B (R3)에서 정의한 주요항(main section)과 XML기술어(descriptors)와의 관계를 Figure 1에 나타냈다. 이 관계도에서 알 수 있듯이 유효한 안전성 보고를 위한 필수 최소정보는 최소한 한 명의 식별가능한 환자(예: 환자의 이니셜, 나이, 성별), 한 명의 식별가능한 보고자(예: 보고자의 이니셜, 주소, 자격), 한 개의 이상반응/이상사례(또는 결과)와 한 개의 의심되는 약물 또는 상호작용 약물이 포함되어야 한다. ICSR 보고에 요구되는 필수 최소정보 이외에도 보고자 관리번호(C.1.1), 보고유형(C.1.3), 가장 최근의 발생인지일(C.1.5), 이 보고가 해당 국가의 신속보고 기준에 해당하는지(C.1.7), 전세계 고유식별(C.1.8), 보고자의 국가코드(C.2.r.3), 발신자의 소속조직(C.3.2), 약물 이상반응/이상사례가 관찰된 연구유형(C.5.4) 등의 특정 행정적인 정보는 보고를 적절하게 처리하기 위해 제공되어야 한다. 실제, 대부분의 ICSR에 있어서 상당수의 데이터 항목은 알 수 없을 수 있다. 따라서 ICSR에 보고 할 때 모든 정보가 전송되지는 않는다. ICSR 정보는 전자적으로 전송되기 때문에 알 수 없거나 선택적인 데이터 항목에 값을 할당할 필요는 없다. 그러나 적용되지 않거나, 모르거나, 개인정보보호법에 의해「보호」되어 데이터 항목이 비어있는 경우도 있을 수 있다. 이 경우 빈 값으로 표현되는 정보에 대해서는 데이터 항목에 관한 메시지가 포함되어야 하며, 데이터 없음 및 그 이유를 나타내야 한다.
ICSR의 정보기술이나 코딩에 사용되는 용어 및 관리 용어는 여러가지가 존재한다. ICSR 메시지에서 사용하는 용어 및 어휘로는 의약품 식별표준(Identification of Medicinal Product, IDMP), 국제의약용어(MedDRA), ICH ICSR용으로 작성된 ICH가 유지하는 코드세트 및 객체 식별자(Object Identifier, OID), 국가코드(ISO 3166 Part I [alpha-2]), 성별표시를 위한 코드(ISO 5218), 언어 표시에 대한 코드(ISO 639-2), 측정단위에 관한 통일코드(The Unified Code for Units of Measures, UCUM) 등의 국제표준코드세트 등이 있다.
E2B (R2)에서 E2B (R3)로 전환
ICH E2B 전문가 워킹그룹은 1997년 E2B 가이드라인을 처음 발표하였고, 2000년 수정된 버전을 발표하였다. 이후 2001년 일부를 수정하여 버전 4.4.1을 발표하였으며, 이 버전은 이어서 E2B (R2) 가이드라인으로 이름이 변경되었다. ICH M2 전문가 워킹그룹은 E2B (R2)의 이행을 위한 메시지 사양으로 ICH ICSR DTD 버전2.1을 개발하였고, 2001년 이 가이드라인은 step 4에 도달하였다. 이 가이드라인에 따라 ICSR의 전자 제출관련 가이드라인은 모든 ICH 회원국(미국, 일본 및 EU 등)에 적용되었고, 각 지역의 현황에 맞추어 구현되었다. ICH E2B (R2)는 ICSR 데이터 요소에 대한 정보 규격을 포함하고 있다. ICH 회원국은 승인된 권고사항을 채택해야 하며, 이를 즉각적으로 채택할 필요는 없다. 하지만 회원국이 전자 전송을 시행할 때에는 일정한 기준이 있어야만 하며, ICH E2B는 ICSR 전송과 이를 구성하는 데이터 요소를 사용하는 방법에 대한 최신 내용을 제공한다.
2011년 국제표준화기구(International Organization for Standards, ISO)와 유럽표준화위원회(European Committee for Standardization, CEN)가 협력하여 의약품 등에 대한 이상사례, 제품문제 및 소비자 불만신고를 지원하는데 필요한 정보수집을 위하여 보고양식 표준인 ISO/HL7 27953을 마련하였다. ISO/HL7 27953은 HL7 버전 3(HL7 V3)를 기반으로 하며, HL7 모델 기반 방법론은 의료 시스템 상호 운용성 및 정보교환을 위한 합의 기반표준을 개발하는데 사용된다. ISO는 상호 운용성 표준 ISO/HL7 Release 3 표준(또는 HL7 ICSR R3)을 준수하기 위한 인증기관으로서 ISO/HL7 27953-2 표준을 준수하고 있다. ISO/HL7 27953은 ISO New Work Item Proposal N545 (ICSR의 구조 및 데이터 요소), HL7 ICSR Release 1의 규범적 표준 및 HL7 Release 2 시험사용을 위한 초안 표준안(Draft Standard for Trial Use, DSTU)을 토대로, 내용과 메시지의 구체화를 더욱 확고히 한다. 이 표준은 2011년 11월 국제표준으로 ISO에 의해 발표되었다[6]. 이후 ICH는 ICH 운영위원회는 그 동안 회원국 내에서 운영되던 E2B (R2) 기술규격서를 표준개발기구(Standards Development Organization, SDO)와 협력을 통해 규제 및 보건분야 전반으로 보다 광범위하게 상호운용이 가능한 E2B (R3)로 개발하였으며, ISO, HL7, CEN 외에도 국제 임상 데이터 교환 표준 컨소시엄(Clinical Data Interchange Standards Consortium, CDISC), 국제보건용어표준개발기구(International Health Terminology Standards Development Organization, IHTSDO), GS1 (Global Standard No.1)이 SDO 세계보건정보처리표준화에 대한 공동 이니셔티브(Joint Initiative on SDO Global Health Informatics Standardization)에 참여하여 공동 표준을 개발하였다. ICH E2B (R3) 메시지 표준은 ISO/HL7 27953-2를 기반으로 개발되었으며, E2B (R3) 보고요건을 충족시키기 위해 보다 제한된 ISO ICSR 표준을 채택하였다. 이 표준은 ‘ICH subset’이지만, ICH E2B (R3) 가이드라인 보다 넓은 범위 및 실무예제에 적용할 수 있다. E2B (R3) 가이드라인에서는 ICH 소관 밖의 ISO/HL7 27953-2 표준요소는 명시되지 않는다. ICH는 이 표준이 E2B (R3)가 지정한 사용 분야를 아우르는 ICH 도입 가이드라인으로 사용되어야 한다고 정의한다. ISO 표준 자체는 ICH에서는 사용되지 않지만 특정지역에서 사용될 수 있는 추가적인 데이터 요소/요건을 포함하고 있다. 이러한 사용은 적절한 경우 지역적 도입 지침으로 정의될 수 있다.
현재 WHO를 비롯한 국외 주요국의 의약품안전성 모니터링 데이터베이스는 대체로 ICH E2B (R2) 가이드라인 및 문서형식의 정의(DTD) 버전 2.1을 기반으로 운용되고 있다. ICH E2B (R3) 가이드라인이 현재의 E2B (R2) 가이드라인과 M2 가이드라인을 개선할 것으로 예상되지만, 모든 이해관계자(규제당국, 제약회사, 의약품사업 분야의 다른 관계자)가 새로운 가이드라인을 이행하여 의약품안전성 모니터링 데이터베이스를 E2B (R3)로 시스템을 구현하는 데까지는 이행기간이 필요하다.
ISO ICSR 표준(ISO/HL7 27953-2:2011)은 ICSR 메시지를 작성하는데 사용되는 기술구조(schema file, 스키마)를 제공한다. ICH E2B (R3) 규격은 ICSR 내용에 대한 데이터 요소를 제공하는 합의문서이며, ISO ICSR의 모든 데이터 요소와 요구사항이 사용된 것은 아니다. 즉, 미국, 유럽 및 일본의 3개 ICH 지역의 요구사항 대부분은 ICH E2B (R3) IG (Implementation Group, 구현그룹)에 의해 통합될 수 있었지만, ICH E2B (R3)에서 다루어지지 않았다는 점이다. 대신 지역 간 법률차이로 인해 발생하는 추가 요구사항은 지역의 ICSR 구현지침(Implementation Guide)에 반영하여 표준인 ICH E2B (R3) 사양을 보완한다. 즉, ICH는 각 지역의 구현 지침을 개발하는 것을 인정하고 있으며, 각 지역은 다른 ICH 지역에서 요구하는 추가 데이터 요소가 포함된 ICSR 메시지를 거부하지는 않도록 한다. ICH E2B (R3) 구현 전문가 그룹(IWG)은 지역 구현지침을 검토하여 해당 지역 구현 간에 충돌이 없는지 확인해야 하며 IWG는 표준 구현자를 돕기 위해 질의응답(Q&A) 문서를 작성하고 있다. 현재 Q&A 문서는 2013년 제정, 2016년 11월 개정되었으며, 자세한 내용은 ESTRI 홈페이지를 통하여 제공되고 있다. E2B (R3) 가이드라인이 발간되었지만 유럽과 미국 등 선진국에서는 이 시스템을 도입하기 위하여 ICH 문서의 핵심 세트에 기초하여 자체 지역 IG를 구성하고, 각 지역의 구현지침을 개발하였다. 또한 R3에 기반한 시스템 구축 및 R2와 R3를 동시에 운영하는 유예기간을 두고 자료의 호환성이나 수집된 자료를 시범분석하여 취약점을 보완하는 등 중장기 계획을 세우고 있다(Table 1)[7-17]. 각국에서는 E2B (R3) 규정이 의무화되기 전에 이해관계자가 안전 데이터베이스를 변경해야 한다는 것을 잘 알고 있으며, E2B (R3)에 부합하려면 기관에서 사전에 프로세스 변경을 계획하고 추진하여 E2B (R2) 및 E2B (R3) 제출을 모두 수행할 준비가 되어 있어야 한다. 그러나 E2B (R2) 데이터베이스를 가지고 E2B (R3)에 메시지를 보내는 것은 불가능하고, E2B (R2)데이터베이스는 E2B (R3) 안전메시지를 수신하여 이를 E2B (R2)로 변환할 수 있게는 해준다. 위에서 설명한 변경사항은 모든 SOP와 절차문서 및 의약품 감시 시스템 마스터 파일(Pharmacovigilance System Master File, PSMF)에 영향을 미치므로 수정되어야 하며, 이 과정에 참여한 모든 담당자들의 재교육이 상당하게 필요할 것이다. 기관에서는 잠재적인 문제를 해결할 수 있도록 시스템 테스트를 미리 계획해야 할 것이다.
BFC (Backwards and Forwards Conversion) 도구는 변경을 준비하는 동안 기관에 도움이 될 수 있으며, ICH E2B (R3)의 호환성 지침(APPENDIX I (B) to the Implementation Guide for Electronic Transmission of Individual Case Safety Reports (ICSRs) Backwards and Forwards Compatibility Recommendations)[18]이 제공되고 있다. 이는 각 버전의 자료 변환과정에서 일부 데이터가 손실되는 경우 등의 사례에 대하여 데이터의 무결성을 유지하고, 변환의 한계가 자세히 설명되어야 하며, E2B community에 의해 유지되는 것을 보장하기 위하여 제공된다.
의견: 국내 적용을 위한 제언
국내에서는 식약처에서 2016년 3월 「개별이상사례전자보고양식[E2B (R3)] 가이드라인」을 발간 및 배포하였다. 앞서 언급한 바와 같이 주요국에서는 가이드라인 발간과 함께 지역 구현지침을 마련하고, E2B (R3)를 적용한 부작용보고시스템을 도입하기 위하여 시스템 구축, R2와 R3를 동시에 운영하는 유예기간을 두고 자료의 호환성이나 수집된 자료를 시범 분석하여 취약점을 보완하는 등 중장기 계획을 세우고 있다. 우리나라도 ICH E2B 가이드라인의 요구사항을 벗어나지 않는 범위 내에서 우리나라 고유의 행정 및 법률 등 체계를 고려하여 지역 세부 이행지침 마련이 필요할 것으로 사료된다. 또한 이제까지 기존 국내 의약품 부작용 보고 시스템을 통해 수집된 ICSR을 E2B (R3) 가이드라인에 따라 제출할 수 있도록 세부 이행사항을 설명하고, 제약회사 및 기타 관계자들이 E2B (R3) 데이터 요소에 부합되게 ICSR을 제출할 수 있도록 구체적인 시행계획을 마련해야 한다.
현재 우리나라에서는 KAERS (Korean Adverse Event Reporting System) 시스템을 통해 국내에서 발생하는 이상사례를 자체 양식에 따라 보고받고 있으며, 2014년 7월부터는 ICH E2B (R2) 양식에 따라 국내 판매되는 의약품이 국외에서 사용된 경우 발생한 이상사례를 별도의 시스템을 통하여 보고받고 있다. 국내의 안전성 정보관리와 관련된 행정절차에 따라 ICH E2B (R3) 가이드라인의 모든 항목을 반영하면서도 우리나라 고유의 자체 추가 데이터 항목이 추가된 지역 구현지침 개발이 필요할 것이다. 현재 국내 규정에 따라 실시되는「재심사제도」와 재평가제도」, 「품목허가갱신제도」, 「DUR 제도」에 대하여 데이터 항목을 신설하여 기재할 수 있도록 하는 것이 바람직할 것으로 사료된다. “신약 등의 재심사 기준(식약처고시)”에서는 신약의 경우에는 3,000례의 조사 대상자에 대한 ICSR을 수집하여 제출하도록 하고 있고, 그 외의 경우 600례의 ICSR을 제출하여 평가하도록 하고 있다. 재평가제도의 경우에도 배합의의(복합제에 한함) 등과 함께 시판 후 부작용 수집사례, 문헌정보 등 안전성 정보를 제출받아 평가하고 있다. 품목허가갱신제도 및 DUR 제도를 운영함에 있어서도 자발적 부작용보고자료는 중요한 역할을 하며, 국제적으로 표준화된 형식으로 제출한다면 효율적인 자료수집 및 분석이 가능할 것으로 생각된다. 이러한 국내 실정을 반영하여 다음과 같은 세부 데이터 요소를 추가하고 관리할 필요가 있다.
용어통합
기존 KAERS에서는 이상사례는 WHO-ART 용어를 사용하고, 의약품적응증, 관련병력, 보고된 사망원인, 부검시 입증된 사망원인, 부모의 과거병력 항목은 KCD-6 용어를 사용하여 개별 이상사례보고를 받아 왔지만 국내에 E2B (R3)를 적용하여 부작용을 보고받기 위해서는 MedDRA용어를 사용해야 한다. 이를 위해 식약처에서는 5년 이내에 MedDRA 가이드라인(안) 및 MedDRA 한글용어집을 마련하고, 공식적으로 한글 MedDRA를 도입할 예정이다. 또한 ICH/MSSO와 협력작업을 통하여 WHO-ART와 MedDRA 변환작업을 추진해야 한다.
G.k 의약품 정보항에 대한 지역관리
의약품정보에 해당하는 IDMP 표준은 규제당국, 제약업체 및 의료전문가가 의약품 데이터를 교환하고 실질적으로 사용할 수 있도록 설계되었다. ICH E2B (R3)는 선택 데이터 요소로 G.k항의 M5 관리용어 세트의 사용을 언급하고 있기 때문에 ISO IDMP 표준의 국내 적용을 위한 추가 연구 및 지원을 위한 정책수립이 필요하다. 국내 의약품에 대한 정보는 식약처 품목 기준코드를 중심으로 관리하고 있으며, IDMP에 해당하는 물질정보, 약제복용량형태, 투여단위, 투여경로 및 포장에 관한 규제정보, 품목에 대한 규제 고유정보, 의약품식별(PhPID)과 측정단위가 허가정보에 대부분 포함되어 있다. 이 정보를 바탕으로 IDMP 표준의 통합을 고려해 볼 수 있다.
국내 상황을 반영한 데이터 요소 추가
식약처는 전체 ICSR 데이터 품질개선 및 기타 식약처 안전성 정보관리 계획을 지원하기 위해 필요한 국내 특정 데이터를 수집한다. 부작용보고 데이터는 E2B (R3) 가이드라인에 따라 XML 스키마 속성이 지원되는 ICSR 파일을 사용하여 수신되며 식약처는 자세한 내용을 제공하기 위해 XML 관찰 속성을 사용하고 주제에 대한 설명을 제공하기 위해 XML 특성을 사용해야 한다. 앞서 언급한 바와 같이, 현재 국내 규정에 따라 실시되는「재심사제도」와「재평가제도」, 「품목허가 갱신제도」, 「DUR 제도」에 대하여 데이터 항목에 대하여 추가 기입할 수 있는 데이터 요소를 표준에 벗어나지 않는 범위 내에서 추가할 수 있을 것이다. 현재 “의약품 등의 안전에 관한 규칙” 별지 제77호의2서식 이상사례·약물 이상반응 보고서의 보고 구분항목에 반영되어 있는 바와 같이 데이터 항목을 추가하는 방안을 제안하고자 한다.
E2B (R3) 가이드라인에서 보고유형 C.1.3 항목에 해당하며, 이 데이터 항목에는 출처에 관계없이 보고의 종류를 입력하는 것이다. 이 항목은 필수항목으로, 1=자발적 보고, 2=시험에서 보고, 3=기타, 4=발신자 정보이용불가(불명)로 입력해야 한다. 문헌정보인 경우에는 C.4항에 문헌정보 출처를 명시하도록 별도의 항목을 마련하고 있기 때문에 여기에 중복 입력하지 않는다. 예를 들어, 문헌보고 중인 사례가 자발적 관찰의 것인 경우는「보고유형」을 자발적 보고로 한다. 문헌보고중인 사례가 시험에서 발생한 경우, 「보고유형」은 시험에서의 보고로 하고, C.5.4항에서 연구의 유형(예: 임상시험 또는 기타)을 구별한다(C.5.4 사용자지침 참조). 문헌보고에서 사례가 자발적으로 관찰되거나 또는 시험에서 발생한 것인지를 알 수 없는 경우 이 항목은 기타로 한다. 발신자 정보이용 불가의 옵션은 첫째 발신자가 보고의 종류를 특정하지 않은 정보를 제2차 발신자(예: 규제당국)가 전송하는 경우에 사용할 수 있다. 이것은 발신자가 보고의 종류를 알고 있지만, 제시된 카테고리에 맞지 않는 것을 나타내는「기타」와는 다르다.
우리나라의 경우는 보고유형을 자발보고, 조사연구, 문헌, 모름, 기타로 크게 구분하고, 조사연구의 경우에는 계획서 번호와 제목을, 재심사 경우에는 사용성적조사, 시판 후 임상연구, 특별조사를 선택하도록 하고, 안전성 정보조사에 의한 연구, 임상연구, 개별 사례연구 및 기타로 입력이 가능하게 세부 항목정보를 수집하고 있다. 보고유형과 조사연구, 임상연구의 경우는 E2B (R3)의 표준서식에 따라 입력하도록 하되, 조사연구정보는 C.5항에 연구제목, 의뢰자 연구번호, 약물 이상반응/이상사례가 관찰된 연구유형, 연구등록번호, 연구등록 국가 등의 정보를 입력하도록 하기 때문에 이 항목을 활용하면 될 것이다. 재심사 경우 별도의 항목을 신설할 필요가 있으며, 이 항목에 대한 입력규칙을 제안하고자 한다(1= 사용성적조사, 2=시판 후 임상연구, 3=특별조사).
결 론
2010년 시판 후의 약품 부작용 정보보고 및 분석을 전담하는 기관으로서 한국의약품안전관리원 설립과 2013년 식품의약품안전처의 허가를 위한 임상시험정보 제출을 위한 CDISC 시스템 도입 및 개발로 IT 기술을 기반으로 한 전주기에 걸친 의약품 안전성 정보를 수집하고 분석하기 위한 기본틀은 일부 갖추어져 있다고 할 수 있다. 다만 국제조화를 위한 가이드라인 및 전자표준 적용과 표준화된 시스템에 자동화된 분석툴을 구현하여 더 효율적이고 과학적 방법론에 기반한 유연성 있는 시스템으로 거듭날 필요가 있다. 그러자면 가장 시급히 해결해야 할 문제는 의약품 개발단계의 임상시험 단계에서부터 시판 후의 약품사용으로 인해 발생되는 의약품 부작용 정보를 수집하기 위하여 E2B 표준의 적용이 필수적이다.
개별 이상사례 보고에 있어서 국제 조화된 표준채택과 구조화된 데이터 확립이 가능하다면 이는 국내 의약품 안전성 정보 관리 개선에 기여할 것이다. 또한 표준화된 데이터 구조의 이상사례 정보를 국내·외 비교분석하면 의약품을 사용하는데 있어 보다 효과적인 커뮤니케이션, 분석, 개선방안을 제공할 수 있다. 한국의 국내 이상사례 정보도 ICH E2B 표준에 따라 보고받는다면 WHO의 전자보고시스템에 효율적으로 연계 및 보고할 수 있도록 표준화된 자료가 되며, 이는 약물감시 분야에서의 국제적 위상을 제고할 수 있을 것이다. 또한 한국형 안전성 정보항목으로 재심사, 재평가 및 DUR 제도와 관련된 정보를 동시에 수집함으로써 국내제도의 효용성 제고에 도움이 될 것이다.
개별 이상사례 보고는 국제표준을 준수해야 하므로 ICH E2B 표준을 따라야 한다는 것은 규제당국 및 제약회사 모두 동의할 것이다. 개별 이상사례 보고의 표준에 대하여 조화가 되지 않으면 보고의 다양성 때문에 보고의 효율성이 떨어지게 되고 보고자의 부담은 늘어난다. 이런 부조화로 인해 국제적으로 개별 이상사례 정보가 일치하지 않을 수 있기 때문에, 전세계적 안전성 정보교환에 많은 규제당국과 제약회사를 고려하여 데이터베이스 간의 직접 전송을 가능하게 하는 표준화된 메시지 형식을 사용해야 한다. ICSR의 전자적 전송은 전세계적 약물감시에서 중요 요소가 된다. 전세계적으로 방대한 양의 데이터와 수많은 정보교환자들을 고려하여 교류가 가능한 데이터베이스에 의해 자동으로 생성 및 처리가 가능한 형식으로 하는 안전성 보고의 전자적 전송을 계속해서 강화할 필요가 있다. 표준화된 데이터의 정보교환은 공통된 하나의 문서만으로도 많은 다른 국가뿐만 아니라 다른 구조의 데이터와도 커뮤니케이션이 가능하게 해 줄 것이다.